
Enllaç C-Li

En química organometàl·lica, un organolític és un compost químic covalent que conté enllaços carboni-liti (C-Li). Són reactius importants en la síntesi orgànica i s'utilitzen amb freqüència per transferir el grup orgànic o l'àtom de liti als substrats en passos sintètics, mitjançant addició nucleòfila o desprotonació simple.[1] Els reactius organolítics s'utilitzen a la indústria com a iniciador per a la polimerització aniònica, que condueix a la producció de diversos elastòmers. També s'han aplicat en síntesi asimètrica en la indústria farmacèutica.[2]
A causa de la gran diferència d'electronegativitat entre l'àtom de carboni i l'àtom de liti, l'enllaç C-Li és molt iònic. Com la naturalesa electropositiva del liti fa que la densitat de càrrega de l'enllaç estigui sobre l'àtom de carboni, es crea un carbanió. Aquesta naturalesa extremadament polar de l'enllaç C-Li fa que els reactius organolítics siguin bons nucleòfils i bases fortes. Per aquesta reactivitat són incompatibles amb aigua (H₂O) oxigen (O₂), i diòxid de carboni (CO₂) i han de ser manejats sota atmosfera protectora com nitrogen, o preferiblement argó.
Per a la síntesi orgànica de laboratori, molts reactius organolítics estan comercialment disponibles en forma de solució. Aquests reactius són altament reactius, i de vegades són pirofòrics.
Història i desenvolupament
[modifica]
Els estudis dels reactius organolítics van començar els anys 1930 i van ser pioners Karl Ziegler, Georg Wittig i Henry Gilman. Aquests químics van trobar que, en comparació amb els reactius de Grignard, els reactius organolítics sovint poden fer les mateixes reaccions amb majors velocitats i majors rendiments, com en el cas de la metal·lació.[3] Des d'aleshores, els reactius organolítics han superat els reactius de Grignard en ús.
La investigació en curs se centra en la naturalesa de l'enllaç carboni-liti, estudis estructurals d'agregats organolítics, reactius quirals organolítics i síntesi asimètrica, i el paper dels reactius organolítics en la preparació de noves espècies organometàl·liques.[4]
Estructura
[modifica]
Encara que les espècies simples d'alquil-liti es representen sovint com un monòmer RLi, també existeixen com agregats (oligòmers) o polímers.[5]
Les seves estructures depenen de la naturalesa del substituent orgànic i la presència d'altres lligands.[6][7] Aquestes estructures han estat dilucidades per una varietat de mètodes, principalment l'espectroscòpia RMN ⁶Li, 7Li i 13C, i l'anàlisi de difracció de raigs X.[1]
La química computacional admet aquestes tasques.[5]
Naturalesa de l'enllaç carboni-liti
[modifica]Les relatives electronegativitats del carboni i el liti suggereixen que l'enllaç C-Li serà altament polar.[8][9][10] Tanmateix, certs compostos organolítics tenen propietats com la solubilitat en dissolvents no polars que compliquen el problema.[8] Encara que la majoria de dades suggereixen que l'enllaç C-Li és essencialment iònic, s'ha debatut si existeix un petit enllaç covalent en l'enllaç C-Li.[9][10]
En els compostos al·lil-litics, el catió de liti es coordina amb la cara de l'enllaç
Estructures d'estat sòlid
[modifica]



Igual que altres espècies consistents en subunitats polars, les espècies organolítics s'agreguen.[7][13] La formació d'agregats està influenciada per interaccions electroestàtiques, la coordinació entre el liti i les molècules solvents circumdants o els additius polars, i els efectes estèrics.[7]
Un bloc bàsic per construir estructures més complexes és un centre de carbanió que interactua amb un triangle Li₃ en una forma

Amides de liti comunes, com per exemple la bis(trimetilsilil)amida de liti (hexametildisilazida de liti / LiHMDS / LiN(SiMe₃)₂) i la diisopropilamida de liti (LDA / [(CH₃)₂CH]₂NLi), també estan subjectes a l'agregació.[14] Les amides de liti adopten estructures de tipus d'escala polimèrica en dissolvent no-coordinant en estat sòlid, i generalment existeixen com dímers en dissolvents etèrics. En presència de lligands fortament donants, es formen centres de trímers o tetràmers de liti.[15] Per exemple, la LDA existeix principalment com a dímer amb el tetrahidrofurà (THF / (CH₂)₄O).[14] Les estructures d'amides de liti comuns, com la diisopropilamida de liti (LDA) i l'hexametildisilazida de liti (LiHMDS), han estat àmpliament estudiades per Collum i companys de treball utilitzant espectroscòpia RMN.[16]
Una altra classe important de reactius són els silil-liti, àmpliament utilitzats en la síntesi de complexos organometàlics i dendrímers de polisilans (Si-Si).[7][17] En estat sòlid, en contrast amb els reactius alquil-liti, la majoria dels silil-liti tendeixen a formar estructures monomèriques coordinades amb molècules de dissolvents com el THF, i només uns pocs silil-liti s'han caracteritzat com a agregats més alts.[7] Aquesta diferència pot sorgir a partir del mètode de preparació dels silil-liti, l'obstacle estèric causat pels substituents alquils voluminosos del silici i la naturalesa menys polaritzada dels enllaços Si-Li. L'addició de lligands fortament donants, com el TMEDA i (-)-esparteïna, pot desplaçar la coordinació de molècules de dissolvents en silil-liti.[7]
Solució de l'estructura
[modifica]Confiar únicament en la informació estructural d'agregats organolítics obtinguts en estat sòlid a partir d'estructures de cristall té certs límits, ja que és possible que els reactius organolítics adoptin diferents estructures en reacció a l'entorn de la solució.[6] A més, en alguns casos, l'estructura cristal·lina d'una espècie d'organolítics pot ser difícil d'aïllar. Per tant, estudiar les estructures dels reactius organolítics, i els intermedis que contenen liti en forma de solució, és summament útil per comprendre la reactivitat d'aquests reactius.[18] L'espectroscòpia RMN ha sorgit com una poderosa eina per a l'estudi dels agregats organolítics en solució. Per a les espècies d'alquil-liti, l'acoblament J de C-Li es pot utilitzar sovint per determinar la quantitat de liti que interactua amb un centre de carbanió i si aquestes interaccions són estàtiques o dinàmiques.[6] Els senyals de RMN separats també poden diferenciar la presència de múltiples agregats des d'una unitat monomèrica comuna.[19]
Les estructures dels compostos organolítics es veuen afectades per la presència de bases de Lewis, com per exemple, el tetrahidrofurà (THF / (CH₂)₄O), el dietilèter (Et₂O / (C₂H₅)₂O), la tetrametiletilendiamina (TMEDA / (CH₃)₂NCH₂CH₂N(CH₃)₂) o l'hexametilfosforamida (HMPA / [(CH₃)₂N]₃PO).[5] El metil-liti és un cas especial, en què la solució amb èter (R-O-R') o amb l'additiu polar HMPA no desagreguen l'estructura tetramèrica en estat sòlid.[7] D'altra banda, el THF desagrega el butil-liti en hexàmer: el tetràmer és l'espècie més important, i
| Solvent | Estructura | |
|---|---|---|
| metil-liti | THF | tetràmer |
| metil-liti | èter/HMPA | tetràmer |
| n-butil-liti | pentà | hexàmer |
| n-butil-liti | èter | tetràmer |
| n-butil-liti | THF | tetràmer-dímer |
| sec-butil-liti | pentà | hexàmer-tetràmer |
| isopropil-liti | pentà | hexàmer-tetràmer |
| terc-butil-liti | pentà | tetràmer |
| terc-butil-liti | THF | monòmer |
| fenil-liti | èter | tetràmer-dímer |
| fenil-liti | èter/HMPA | dímer |
Estructura i reactivitat
[modifica]A mesura que les estructures dels reactius organolítics canvien segons el seu entorn químic, també ho fan la seva reactivitat i selectivitat.[7][21] Una pregunta sobre la relació estructura-reactivitat és si existeix una correlació entre el grau d'agregació i la reactivitat dels reactius organolítics. Es va proposar originalment que els agregats més baixos com els monòmers són més reactius en els alquil-liti.[22] Tanmateix, també s'han descobert vies de reacció en què els dímers o altres oligòmers són espècies reactives,[23] i per a les amides de liti com la LDA, les reaccions basades en dímers són comunes.[24] Una sèrie d'estudis cinètics de solució de reaccions mitjanes de la LDA suggereixen que els agregats més baixos dels enolats no condueixen necessàriament a una major reactivitat.[16] A més, algunes bases de Lewis incrementen la reactivitat dels compostos organolítics.[25][26]
No obstant això, si aquests additius funcionen com a lligands quelants forts, i com l'augment observat de la reactivitat es relaciona amb els canvis estructurals dels agregats causats per aquests additius, no sempre són clars.[25][26] Per exemple, la TMEDA augmenta la velocitat i l'eficiència en moltes reaccions que impliquen reactius organolítics.[7] Pel que fa als reactius alquil-liti, la TMEDA funciona com a lligant donant,[5] redueix el grau d'agregació i augmenta la nucleofilicitat d'aquestes espècies.[27] Tanmateix, la TMEDA no sempre funciona com un lligant donant al catió de liti, especialment en la presència d'un centre d'oxigen i nitrogen aniònic. Per exemple, només interactua dèbilment amb la LDA i la LiHMDS fins i tot en dissolvents d'hidrocarburs sense lligands de donants competidors.[28] En la litiació, mentre que el THF actua com un fort lligant de donació a la LiHMDS, la TMEDA feblement coordinadora es dissocia fàcilment des de la LiHMDS, donant lloc a la formació de dímers de LiHMDS que és l'espècie més reactiva. Per tant, en el cas de la LiHMDS, la TMEDA no augmenta la reactivitat reduint l'estat d'agregació.[29] A més, a diferència dels compostos d'alquil-liti simples, la TMEDA no desagrega l'acetofenolat de liti en la solució de THF.[6][30] L'addició d'HMPA a amides de liti, com la LiHMDS i la LDA, sovint dona lloc a una barreja d'agregats dímers / monòmers en THF. Tanmateix, la relació entre l'espècie dímer / monòmer no canvia amb una major concentració d'HMPA, per tant, l'augment observat de la reactivitat no és el resultat de la desagregació. El mecanisme de com aquests additius augmenten la reactivitat encara s'està investigant.[21]
Reactivitat i aplicacions
[modifica]L'enllaç C-Li en els reactius organolítics està molt polaritzat. Com a resultat, el carboni atrau la major part de la densitat electrònica en l'enllaç i s'assembla a un carbanió. Així, els reactius organolítics són fortament bàsics i nucleofílics. Algunes de les aplicacions més comunes dels reactius organolítics en la síntesi inclouen el seu ús com nucleòfils, bases fortes de desprotonació, iniciador per polimerització i material de partida per a la preparació d'altres compostos organometàlics.
Reactius organolítics com nucleòfils
[modifica]Reaccions de carbolitiació
[modifica]Com a nucleòfils, els reactius organolítics es sotmeten a reaccions de carbolitiació, on l'enllaç de carboni-liti s'afegeix a través d'un enllaç doble o triple de carboni-carboni, formant noves espècies organolítiques.[31] Aquesta reacció és la reacció més emprada dels compostos organolítics. La carbolitiació és clau en els processos de polimerització aniònica, i el n-butil-liti s'utilitza com a catalitzador per iniciar la polimerització d'estirè (C₆H₅CH=CH₂), butadiè (C₄H₄) o isoprè (2-metil-1,3-butadiè / CH₂=C(CH₃)CH=CH₂) o les seves mescles.[32][33]

Una altra aplicació que aprofita aquesta reactivitat és la formació de compostos carbocíclics i heterocíclics mitjançant la carbolitiació intramolecular.[31] Com una forma de ciclació aniònica, les reaccions de carbolitiació intramolecular ofereixen diversos avantatges respecte a la ciclació radical. En primer lloc, és possible que l'espècie d'organoliti cíclic del producte reaccioni amb electròfils, mentre que sovint és difícil atrapar un radical intermediari de l'estructura corresponent. En segon lloc, les ciclacions aniòniques són sovint més estereoespecífices que la ciclació radical, particularment en el cas del 5-hexenil-liti (C₆H11Li). La carbolitiació intramolecular permet l'addició del alquil-liti i vinil-liti (C₂H₃Li) als enllaços triple i mono-alquil i substituir els dobles enllaços. Els aril-liti també es poden sotmetre a l'addicció si es forma un anell de 5 membres. Les limitacions de la carbolitiació intramolecular inclouen la dificultat de formar anells de 3 o 4 membres, ja que les espècies intermèdies d'organolítics cíclic solen patir obertures anulars.[31]
A continuació es mostra un exemple de reacció de carbolitiació intramolecular. Les espècies de liti derivades de l'intercanvi de liti-halògens canvien cíclicament per formar el vinil-liti a través del tancament de l'anell 5-exo-trig. Les espècies de vinil-liti reaccionen amb els electròfils i produeixen compostos funcionalitzats de ciclopentilidè .[34]

Addició als compostos carbonílics
[modifica]Els reactius organolítics nucleòfils poden afegir-se a dobles enllaços carbonil electrofílics per formar enllaços de carboni-carboni. Poden reaccionar amb aldehids i cetones per produir alcohols. L'addició procedeix principalment a través de l'addició polar, en què l'espècie organolítica nucleòfila ataca des de la direcció equatorial i produeix l'alcohol axial.[35] L'addició de sals de liti com LiClO₄ pot millorar l'estereoselectivitat de la reacció.[36]

Quan la cetona està estèricament impedida, l'ús de reactius de Grignard condueix sovint a la reducció del grup carbonil en comptes d'afegir-lo.[35] No obstant això, els reactius d'alquil-liti tenen menys probabilitats de reduir la cetona, i es poden utilitzar per sintetitzar alcohols substituïts.[37]
A continuació es mostra un exemple d'addició d'etil-liti amb adamantona per produir alcohol terciari.[38]

Els reactius organolítics també són superiors als reactius de Grignard en la seva capacitat de reacció amb àcids carboxílics (–COOH) per formar cetones.[35] Aquesta reacció es pot optimitzar controlant acuradament la quantitat d'addició de reactius organolítics, o utilitzant clorur de trimetilsilil (TMSCl / Me₃SiCl / (CH₃)₃SiC) per extingir l'excés de reactiu de liti.[39] Una manera més comuna de sintetitzar cetones és mitjançant l'addició de reactius d'organolític a amides de Weinreb (N-metoxi-N-metilamides). Aquesta reacció proporciona cetones quan els reactius organolítics s'utilitzen en excés, a causa de la quelació de l'ió de liti entre l'oxigen del N-metoxi i l'oxigen del grup carbonil, que forma un intermedi tetraèdric que es col·lapsa sota el treball de l'àcid.[40]

Els reactius organolítics també poden reaccionar amb el diòxid de carboni (CO₂) per formar àcids carboxílics.[41]
En el cas dels substrats d'enona, on són possibles dos llocs d'addició nucleòfila (addició 1,2 al carboni carbonil, o addició conjugada 1,4 al

Els reactius organolítics també poden realitzar addició nucleòfila enantioselectiva al carbonil i als seus derivats, sovint en presència de lligands quirals. Aquesta reactivitat s'aplica àmpliament en la síntesi industrial de compostos farmacèutics. Un exemple és la síntesi de Merck i Dupont de l'Efavirenz (EFV), un potent inhibidor de la transcriptasa inversa del VIH. L'acetilur de liti (LiC≡CH · NH₂CH₂CH₂NH₂) s'agrega a una cetona pro-quiral per produir un producte alcohòlic quiral. L'estructura de l'intermedi de la reacció activa es va determinar mitjançant estudis d'espectroscòpia RMN en estat de solució i cristal·lografia de raigs X de l'estat sòlid per ser un tetràmer cúbic 2:2.[44]

Reaccions del tipus SN2
[modifica]Els reactius organolítics poden servir com nucleòfils i dur a terme reaccions de tipus SN2 amb halurs alquils o al·lils.[45] Encara que es consideren més reactius que les reaccions de Grignard en l'alquilació, el seu ús encara està limitat a causa de reaccions secundàries que competeixen, com ara reaccions radicals o intercanvi de metall-halogen. La majoria dels reactius organolítics utilitzats en les alquilacions de SN2 són més estabilitzats, menys bàsics i menys agregats, com heteroàtoms estabilitzats de reactius aril-liti o al·lil-liti.[6] S'ha demostrat que l'HMPA augmenta la velocitat de reacció i els rendiments del producte, i la reactivitat dels reactius aril-liti sovint es veu reforçada per l'addició d'alcòxids de potassi.[35] Els reactius organolítics també poden realitzar atacs nucleòfils amb epòxids per formar alcohols.

Reactius organolítics com a base
[modifica]Els reactius organolítics proporcionen una àmplia gamma de basicitat. El terc-butil-liti, amb tres grups alquils donants feblement electrònics, és la base més sòlida disponible comercialment (pKa = 53). Com a resultat, els protons àcids a -OH, -NH i -SH són sovint protegits en presència de reactius organolítics. Algunes de les bases de liti que s'utilitzen habitualment són espècies d'alquil-lití com el n-butil-liti i les dialquilamides de liti (LiNR₂). Els reactius amb grups R voluminosos com la diisopropilamida de liti (LDA) i el bis(trimetilsilil)amida de liti (LiHMDS) sovint són estèricament impedits per l'addició nucleòfila i, per tant, són més selectius per a la deprotonació. Les dialquilamides de liti (LiNR2) són àmpliament utilitzades en la formació d'enolats i la reacció aldòlica.[46] La reactivitat i la selectivitat d'aquestes bases també estan influenciades per dissolvents i altres ions en contra.
Metal·lació
[modifica]La metal·lació amb reactius d'organolític, també coneguda com a «litiació» o «intercanvi liti-hidrogen», s'aconsegueix quan un reactiu organolític, més comunament un alquil-liti, absorbeix un protó i forma una nova espècie d'organolític.
-
()

Els reactius més comuns de les metal·lacions són els butil-liti. Generalment, el terc-butil-liti i sec-butil-liti són els més reactius i tenen una millor selectivitat que el n-butil-liti, però, a més, són més cars i difícils de manejar.[46] La metal·lació és una manera comuna de preparar reactius organolítics versàtils. La posició de la metal·lació està majoritàriament controlada per l'acidesa de l'enllaç C-H. Sovint la litiació es produeix en una posició

Directed ortho metalation

L'orto-metal·lació dirigida (DoM) és una eina important en la síntesi de compostos aromàtics substituïts de regions específiques. Aquest enfocament de la litiació i la posterior extinció de les espècies intermèdia de liti amb electròfils sovint és superior a la substitució electrofílica aromàtica a causa de la seva alta regioselectivitat. Aquesta reacció avança a través de la deprotonació per reactius organolítics en les posicions
Superbases
[modifica]La incorporació d'alcòxid de potassi a l'alquil-liti augmenta considerablement la basicitat de les espècies organolítiques.[50] La «superbase» més comuna es pot formar mitjançant l'addició de t-butòxid de potassi (KOtBu / C₄H9KO) al n-butil-liti (n-BuLi), sovint abreujat com a reactius «LiCKOR». Aquestes superbases són altament reactives i, sovint, reactives estereoselectives. A l'exemple següent, la base de LiCKOR genera una espècie esteroespecifica de crotilboronat a través de la metal·lació i posterior intercanvi de liti-metaloide.[51]
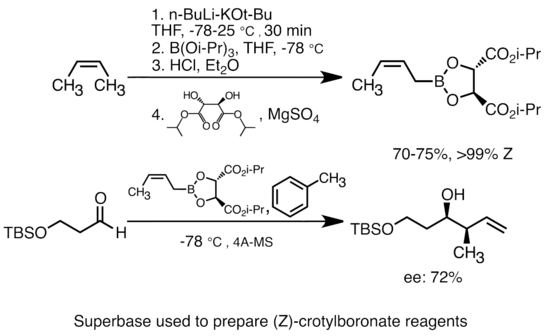
Superbase;

Metal·lació asimètrica
[modifica]Es poden obtenir espècies d'organoliti enantiòmerament enriquides a partir de la metal·lació asimètrica de substrats proquirals. La inducció asimètrica requereix la presència d'un lligand quiral com la (-)-esparteïna.[48] La relació enantiomèrica de l'espècie de liti quiral sovint es veu influenciada per les diferències en les taxes de deprotonació. A l'exemple següent, el tractament de N-Boc-N-bencilamina amb n-butil-liti en presència de (-)-esparteïna proporciona un enantiòmer del producte amb un elevat excés enantiomèric. La transmetal·lació amb clorur de trimetilestany (Me₃SnCl / (CH₃)₃SnCl) proporciona l'enantiòmer contrari.[52]
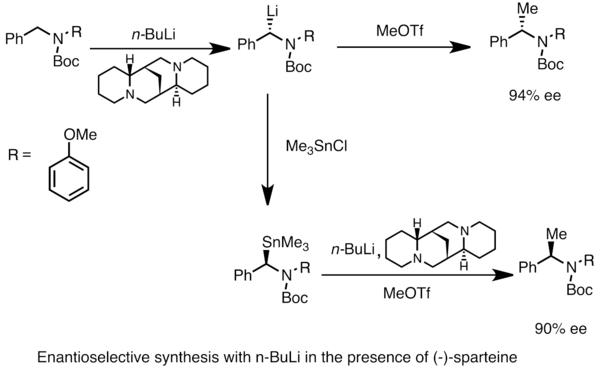
Asymmetric synthesis with nBuLi and (-)-sparteine
-sparteine.png)
Formació d'enolats
[modifica]Els enolats de liti es formen mitjançant la desprotonació d'un enllaç C-H

Sample aldol reaction with lithium enolate

La formació d'enolat de liti es pot generalitzar com una reacció àcid-base, en què el protó
L'estereoquímica i el mecanisme de formació de l'enolat han guanyat molt interès en la comunitat química. Molts factors influeixen en el resultat de l'estereoquímica de l'enolat, com ara efectes estèrics, dissolvents, additius polars i tipus de bases d'organolític. Entre els molts models utilitzats per explicar i predir la selectivitat en estereoquímica dels enolats de liti es troba el model d'Irlanda.[54]
En aquest supòsit, un LDA monomèric reacciona amb el substrat de carbonil i forma un estat de transició de tipus Zimmerman-Traxler cíclic. El (E) enolat està afavorit a causa d'una interacció desfavorable de sin-pentà en l'estat de transició (Z) enolat.[53]

Ireland model for lithium enolate stereoselectivity. In this example, the (E) enolate is favored,

La incorporació d'additius polars, com ara HMPA o DMPU, afavoreix la formació de (Z) enolats. El model d'Irlanda argumenta que aquests lligands de donants es coordinin amb els cations de liti, de manera que es redueix l'intercanvi de carbonils d'oxigen i liti i l'estat de transició no és tan estretament vinculat com una cadira de sis membres. El percentatge de (Z) enolats també augmenta quan s'utilitzen bases de liti amb cadenes laterals més voluminoses (com ara LiHMDS).[53] No obstant això, encara s'està debatent el mecanisme de com aquests additius inverteixen l'estereoselectivitat.
Hi ha hagut alguns reptes al model d'Irlanda, ja que representa l'espècie de liti com un monòmer en l'estat de transició. En realitat, sovint s'observa una varietat d'agregats de liti en solucions d'enolats de liti i, depenent de substrats específics, dissolvents i condicions de reacció, pot ser difícil determinar quin agregat és l'espècie reactiva real en solució.[53]
Intercanvi halogen-liti
[modifica]L'intercanvi d'halogens amb el liti és una reacció de metàtesi entre una espècie organohalur i una d'organolítica. Gilman i Wittig van descobrir de forma independent aquest mètode a la fi dels anys trenta.[55]
-
()

Encara es debat el mecanisme d'intercanvi halogen-liti.[56] Una via possible implica un mecanisme nucleòfil que genera un «complex at» reversible. Farnham i Calabrese van poder aïllar el «complex at» bis(pentafluorofenil)iodonat de liti amb TMEDA i obtenir la estructura del cristall amb raigs X.[57] El «complex at» reacciona més amb els electròfils i proporciona iodur de pentafluorofenil i C₆H₅Li.[57] Una sèrie d'estudis cinètics també donen suport a una via nucleofílica en la qual el carbanió de l'espècie de liti ataca l'àtom dels halògens sobre l'halur d'aril.[58]
Un altre mecanisme possible implica la transferència d'un únic electró i la generació de radicals. En reaccions d'alquil-liti i halurs d'aril secundaris i terciaris s'han detectat espècies radicals per espectroscòpia RPE.[59] Els estudis mecanicistes de l'intercanvi d'halogen-liti també es compliquen per la formació d'agregats d'espècies d'organolítics.

La taxa d'intercanvi halogen-liti és extremadament ràpida. Normalment és més ràpida que l'addició nucleòfila i de vegades pot superar la taxa de transferència de protons. A l'exemple següent, l'intercanvi entre liti i iodur primari és gairebé instantani, i es descompta la transferència de protons des de metanol (CH₃OH) a terc-butil-liti. El major productealquè es forma en més del 90% de rendiment.[60]

L'intercanvi halogen-liti és molt útil en la preparació de nous reactius organolítics. Els tipus de canvi solen seguir la tendència I > Br > Cl. L'alquilfluorur i l'arilfluorur generalment no són reactius vers als reactius organolítics. L'intercanvi halogen-liti està controlat cinèticament, i la taxa d'intercanvi està influenciada principalment per les estabilitats dels intermedis de carbanió (sp > sp2 > sp3) dels reactius organolítics.[35] [47] Per exemple, els reactius organolítics terciaris més bàsics (generalment n-butil-liti, sec-butil-liti o terc-butil-liti) són els més reactius, i reaccionaran amb alquil-halurs primaris (generalment bromur o iodur) per formar espècies d'organolítics més estable. Per tant, l'intercanvi halogen-liti s'utilitza amb més freqüència per a la preparació de reactius de vinil-liti, aril-liti i alquil-liti primaris. També es facilita l'intercanvi halogen-liti quan hi ha grups alcoxi o heteroàtoms per estabilitzar el carbanió i aquest mètode és especialment útil per a la preparació de reactius funcionals de liti que no poden tolerar les condicions més dures requerides per a la reducció amb el liti metàl·lic.[47]
Els substrats com els halurs de vinil solen sofrir intercanvis halogen-liti amb la retenció estereoquímica del doble enllaç.[61]

Retention of stereochem

A continuació es mostra un exemple de l'ús de l'intercanvi halogen-liti en la síntesi de la morfina (C17H19NO₃). Aquí, el n-butil-liti s'utilitza per realitzar intercanvi halogen-liti amb bromur. El centre nucleòfil del carbanió passa ràpidament a la carbolitiació del doble enllaç, generant un anió estabilitzat pel grup sulfonil adjacent. Una reacció SN2 intramolecular per l'anió forma la columna vertebral cíclica de la morfina.[62]

L'intercanvi halogen-liti és una part crucial de la ciclació Parham.[63] En aquesta reacció, un halur d'aril (generalment ariliodur o arilbromur) s'intercanvia amb l'organolític per formar una espècie d'arè litiat. Si l'arè porta una cadena lateral amb un grup electrofílic, el carbanió unit al liti realitzarà un atac nucleòfil intramolecular i ciclarà. Aquesta reacció és una estratègia útil per a la formació d'heterocicles.[64] A l'exemple següent, s'utilitza la ciclació de Parham en la ciclació d'un isocianat per formar isoindolinona (C₈H9N) que després es converteix en una nitrona (R1R₂C=NR₃+O− ). L'espècie nitrona reacciona amb els radicals i es pot utilitzar com a trampa de radicals (spin trap) per estudiar els processos dels radicals biològics.[65]

Parham cyclization in MitoSpin

Transmetal·lació
[modifica]Els reactius organolítics s'utilitzen sovint per preparar altres compostos organometàl·lics a través de la transmetal·lació. Els compostos d'organocoure, organoestany, organosilici, organoborà, organofosforat, organoceri i organosulfurat es preparen freqüentment mitjançant la reacció de reactius organolítics amb electròfils adequats.
-
()

Els tipus comuns de transmetal·lació inclouen l'intercanvi Li/Sn, Li/Hg, i Li/Te, que són ràpids a baixa temperatura.[46] L'avantatge de l'intercanvi Li/Sn és que els precursors del tri-alquil-estany pateixen poques reaccions laterals perquè els subproductes n-Bu₃Sn resultants no són reactius cap als reactius d'alquil-liti.[46] En el següent exemple, el vinilestany (C₂H₆Sn), sintetitzat a partir d'un terminal alquí, forma vinil-liti través de la transmetal·lació amb n-BuLi.[66]
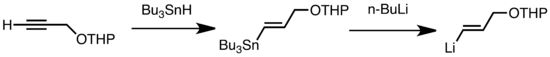
Li Sn exchange

També es pot utilitzar reactius organolítics per preparar compostos d'organozinc mitjançant la transmetal·lació amb sals de zinc.[67]

Organozinc reagents from alkyllithium

Els diorganocuprats de liti es poden formar reaccionant espècies d'alquils de liti amb halur de coure (I). Els organocuprats resultants són generalment menys reactius cap als aldehids i cetones que els reactius organolítics o els reactius de Grignard.[68]
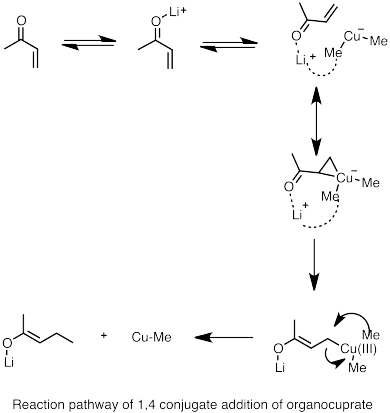
1,4 cuprate addition

Preparació
[modifica]La majoria dels reactius d'alquil-liti simples i amides de liti comunes estan disponibles comercialment en una varietat de dissolvents i concentracions. També es poden preparar reactius organolítics al laboratori. A continuació es detallen alguns mètodes comuns per a la preparació de reactius organolítics.
Reacció amb liti metàl·lic
[modifica]La reducció de l'halur d'alquil amb el liti metàl·lic pot permetre reactius simples d'alquil i aril organolítics.[35]
-
()

La preparació industrial dels reactius organolítics s'aconsegueix utilitzant aquest mètode tractant el clorur d'alquil amb liti metàl·lic que conté 0,5-2% de sodi. La conversió és altament exotérmica. El sodi inicia la via radical i augmenta la velocitat.[69] La reducció es produeix a través d'un camí radical. A vegades, s'utilitza el metall de liti en forma de pols fi en la reacció amb certs catalitzadors com el naftalè (biciclo[4.4.0]deca-1,3,5,7,9-pentè / C10H₈) o el 4,4'-di-t-butilbifenil (DTBB / (CH₃)₃CC₆H₄C₆H₄C(CH₃)₃). A continuació es mostra un exemple de la preparació d'un reactiu de liti funcionalitzat que utilitza la reducció amb el liti metàl·lic:[70]

Reduction with li metal

Un altre substrat que es pot reduir amb liti metàl·lic per generar reactius d'alquil-liti són els sulfurs. La reducció de sulfurs és útil en la formació de reactius organolítics funcionalitzats, com èsters alfa-liti, sulfurs i silans.[71]
Metal·lació
[modifica]Un segon mètode de preparació de reactius organolítics és a través de la metal·lació (intercanvi d'hidrogen-liti). L'acidesa relativa dels àtoms d'hidrogen controla la posició de la litiació.
Aquest és el mètode més comú per a la preparació de reactius d'alquil-liti, ja que el terminal hidrogen lligat al carboni sp és molt àcid i es deprotona fàcilment.[35] Per a compostos aromàtics, la posició de la litiació també es determina per l'efecte directiu dels grups substituents.[72] Alguns dels grups de substitució dirigents més efectius són alcoxí, amido, sulfòxid i sulfonil. Sovint la metal·lació es produeix en la posició orto a aquests substituents. En compostos heteroaromàtics, la metal·lació sol presentar-se en la posició orto a l'heteroàtom.[35][72]

Intercanvi halogen-liti
[modifica]Un tercer mètode per preparar reactius organolítics és a través de l'intercanvi de halogen-liti.
El t-butil-liti o el n-butil-liti són els reactius més utilitzats per generar noves espècies organolítiques mitjançant l'intercanvi halogen-liti. L'intercanvi halogen-liti s'utilitza principalment per convertir els aril i alquenil iodur i bromur amb carbonis sp2 en els compostos organolítics corresponents. La reacció és extremadament ràpida i sol passar de -60 °C fins a -120 °C.[47]
Transmetal·lació
[modifica]El quart mètode per preparar reactius organolítics és a través de la transmetal·lació. Aquest mètode es pot utilitzar per preparar el vinil-liti.
Reacció Shapiro
[modifica]En la reacció de Shapiro, dos equivalents de base forta d'alquil-liti reaccionen amb compostos de p-tosilhidrazona per produir el vinil-liti o, després de sofrir, el producte d'olefina.
Maneig
[modifica]Els compostos organolítics són espècies altament reactives i requereixen tècniques de manipulació especialitzades. Sovint són corrosius, inflamables i de vegades pirofòrics (ignició espontània quan estan exposats a l'oxigen o la humitat).[73] Els reactius alquil-liti també poden experimentar una descomposició tèrmica per formar les corresponents espècies d'alquil i hidrur de liti.[74] Normalment, els reactius organolítics es troben emmagatzemats a -10 °C. Les reaccions es realitzen utilitzant tècniques d'aire lliure.[73] La concentració de reactius d'alquil-liti és determinada sovint per valoració química.[75]
Els reactius organolítics reaccionen amb èters, que sovint s'utilitzen com a dissolvents:[76]
| Solvent / Temp | n-BuLi | s-BuLi | t-BuLi | MeLi | CH₂=C(OEt)-Li | CH₂=C(SiMe₃)-Li |
|---|---|---|---|---|---|---|
| THF / -20 °C | 40 min, 360 min | |||||
| THF / 20 °C | >15 h | 17 h | ||||
| THF / 35 °C | 10 min | |||||
| THF / TMEDA / -20 °C | 55 h | |||||
| THF / TMEDA / 0 °C | 340 min | |||||
| THF / TMEDA /20 °C | 40 min | |||||
| Èter / -20 °C | 480 min | |||||
| Èter / 0 °C | 61 min | |||||
| Èter / 20 °C | 153 h | <30 min | 17 dies | |||
| Èter / 35 °C | 31 h | |||||
| Èter / TMEDA / 20 °C | 603 min | |||||
| DME / -70 °C | 120 min | 11 min | ||||
| DME / -20 °C | 110 min | 2 min | <<2 min | |||
| DME / 0 °C | 6 min |
Referències
[modifica]- ↑ 1,0 1,1 Zabicky, Jacob. «Analytical aspects of organolithium compounds». A: PATAI'S Chemistry of Functional Groups.. John Wiley & Sons, Ltd, 2009. DOI 10.1002/9780470682531.pat0304. ISBN 9780470682531.
- ↑ Wu, G.; Huang, M. «Organolithium Reagents in Pharmaceutical Asymmetric Processes». Chem. Rev., 106, 2006, pàg. 2596–2616. DOI: 10.1021/cr040694k. PMID: 16836294.
- ↑ Eisch, John J. «Henry Gilman: American Pioneer in the Rise of Organometallic Chemistry in Modern Science and Technology†». Organometallics, 21, 25, 2002, pàg. 5439–5463. DOI: 10.1021/om0109408. ISSN: 0276-7333.
- ↑ Rappoport, Z.. The Chemistry of Organolithium Compounds (2 parts).. John Wiley & Sons, Ltd, 2004. ISBN 978-0-470-84339-0.
- ↑ 5,0 5,1 5,2 5,3 5,4 5,5 5,6 5,7 5,8 Stey, Thomas; Stalke, Dietmar. «Lead structures in lithium organic chemistry». A: PATAI'S Chemistry of Functional Groups.. John Wiley & Sons, Ltd, 2009. DOI 10.1002/9780470682531.pat0298. ISBN 9780470682531.
- ↑ 6,00 6,01 6,02 6,03 6,04 6,05 6,06 6,07 6,08 6,09 Reich, Hans J. «Role of Organolithium Aggregates and Mixed Aggregates in Organolithium Mechanisms». Chemical Reviews, 113, 2013, pàg. 7130–7178. DOI: 10.1021/cr400187u. PMID: 23941648.
- ↑ 7,00 7,01 7,02 7,03 7,04 7,05 7,06 7,07 7,08 7,09 Strohmann, C; etal «Structure Formation Principles and Reactivity of Organolithium Compounds.». Chem. Eur. J., 15, 2009, pàg. 3320–3334. DOI: 10.1002/chem.200900041.
- ↑ 8,0 8,1 Jemmis, E.D.; Gopakumar, G. «Theoretical studies in organolithium chemistry». A: PATAI'S Chemistry of Functional Groups.. John Wiley & Sons, Ltd, 2009. DOI 10.1002/9780470682531.pat0297. ISBN 9780470682531.
- ↑ 9,0 9,1 Streiwieser, A. «Perspectives on Computational Organic Chemistry». J. Org. Chem., 74, 2009, pàg. 4433–4446. DOI: 10.1021/jo900497s.
- ↑ 10,0 10,1 Bickelhaupt, F. M.; etal «Covalency in Highly Polar Bonds. Structure and Bonding of Methylalkalimetal Oligomers (CH3M)n (M = Li−Rb; n = 1, 4)». J. Chem. Theory Comput., 2, 2006, pàg. 965–980. DOI: 10.1021/ct050333s.
- ↑ Fraenkel, G.; Qiu, Fayang «Observation of a Partially Delocalized Allylic Lithium and the Dynamics of Its 1,3 Lithium Sigmatropic Shift». J. Am. Chem. Soc., 118, 1996, pàg. 5828–5829. DOI: 10.1021/ja960440j.
- ↑ Fraenkel. G; etal «The carbon-lithium bond in monomeric arllithium: Dynamics of exchange, relaxation and rotation». J. Am. Chem. Soc., 117, 1995, pàg. 6300–6307. DOI: 10.1021/ja00128a020.
- ↑ Power, P.P; Hope H. «Isolation and crystal structures of the halide-free and halide-rich phenyllithium etherate complexes [(PhLi.Et2O)4 and [(PhLi.Et2O)3.LiBr].]». JACS, 105, 1983, pàg. 5320–5324. DOI: 10.1021/ja00354a022.
- ↑ 14,0 14,1 «Synthesis, isolation, and structure of an LDA-THF complex». Journal of Organic Chemistry, 58, 1, 1993, pàg. 1–3. DOI: 10.1021/jo00053a001.
- ↑ Hilmersson, Goran; Granander, Johan. «Structure and dynamics of chiral lithium amides». A: PATAI'S Chemistry of Functional Groups.. John Wiley & Sons, Ltd, 2009. DOI 10.1002/9780470682531.pat0342. ISBN 9780470682531.
- ↑ 16,0 16,1 Collum, D.B.; etal «Lithium Diisopropylamide: Solution Kinetics and Implications for Organic Synthesis». Angew. Chem. Int. Ed., 49, 2007, pàg. 3002–3017. DOI: 10.1002/anie.200603038.
- ↑ Sekiguchi, Akira.; etal «Lithiosilanes and their application to the synthesis of polysilane dendrimers». Coord. Chem. Rev., 210, 2000, pàg. 11–45. DOI: 10.1016/S0010-8545(00)00315-5.
- ↑ Collum, D. B.; etal «Solution Structures of Lithium Enolates, Phenolates, Carboxylates, and Alkoxides in the Presence of N,N,N′,N′-Tetramethylethylenediamine: A Prevalence of Cyclic Dimers». J. Org. Chem., 73, 2008, pàg. 7743–7747. DOI: 10.1021/jo801532d.
- ↑ Reich, H. J.; etal «Aggregation and reactivity of phenyllithium solutions». J. Am. Chem. Soc., 120, 1998, pàg. 7201–7210. DOI: 10.1021/ja980684z.
- ↑ McGarrity, J. F.; Ogle, C.A. «High-field proton NMR study of the aggregation and complexation of n-butyllithium in tetrahydrofuran». J. Am. Chem. Soc., 107, 1985, pàg. 1805–1810. DOI: 10.1021/ja00293a001.
- ↑ 21,0 21,1 Reich, H. J. «What's going on with these lithium reagents». J. Org. Chem., 77, 2012, pàg. 5471–5491. DOI: 10.1021/jo3005155.
- ↑ Wardell, J.L.. «Chapter 2». A: Wilinson, G.. Comprehensive Organometallic Chemistry, Vol. 1. 1st. Nova York: Pergamon, 1982. ISBN 0080406084.
- ↑ Strohmann, C.,; Gessner, V.H. «Crystal Structures of n-BuLi Adducts with (R,R)-TMCDA and the Consequences for the Deprotonation of Benzene». J. Am. Chem. Soc., 130, 2008, pàg. 11719–11725. DOI: 10.1021/ja8017187. PMID: 18686951.
- ↑ Collum, D. B.; etal «Lithium Diisopropylamide: Solution Kinetics and Implications for Organic Synthesis». Angew. Chem. Int. Ed., 46, 2007, pàg. 3002–3017. DOI: 10.1002/anie.200603038.
- ↑ 25,0 25,1 Chalk, A.J; Hoogeboom, T.J «Ring metalation of toluene by butyllithium in the presence of N,N,N′,N′-tetramethylethylenediamine». J. Organomet. Chem, 11, 1968, pàg. 615–618. DOI: 10.1016/0022-328x(68)80091-9.
- ↑ 26,0 26,1 Reich, H.J; Green, D.P «Spectroscopic and Reactivity Studies of Lithium Reagent - HMPA Complexes». JACS, 111, 1989, pàg. 8729–8731. DOI: 10.1021/ja00205a030.
- ↑ Williard, P.G; Nichols, M.A «Solid-state structures of n-butyllithium-TMEDA, -THF, and -DME complexes». JACS, 115, 1993, pàg. 1568–1572. DOI: 10.1021/ja00057a050.
- ↑ Collum, D.B. «Is N,N,N,N-Tetramethylethylenediamine a Good Ligand for Lithium?». Acc. Chem. Res., 25, 1992, pàg. 448–454. DOI: 10.1021/ar00022a003.
- ↑ Bernstein, M.P.; Collum, D.B. «Solvent- and substrate-dependent rates of imine metalations by lithium diisopropylamide: understanding the mechanisms underlying krel». J. Am. Chem. Soc., 115, 1993, pàg. 8008–8010. DOI: 10.1021/ja00071a011.
- ↑ Seebach, D «Structure and Reactivity of Lithium Enolates. From Pinacolone to Selective C-Alkylations of Peptides. Difficulties and Opportunities Afforded by Complex Structures.». Angew. Chem. Int. Ed., 27, 1988, pàg. 1624–1654. DOI: 10.1002/anie.198816241.
- ↑ 31,0 31,1 31,2 Fananas, Francisco; Sanz, Roberto. «Intramolecular carbolithiation reactions». A: PATAI'S Chemistry of Functional Groups.. John Wiley & Sons, Ltd, 2009. DOI 10.1002/9780470682531.pat0341. ISBN 9780470682531.
- ↑ Heinz-Dieter Brandt, Wolfgang Nentwig1, Nicola Rooney, Ronald T. LaFlair, Ute U. Wolf, John Duffy, Judit E. Puskas, Gabor Kaszas, Mark Drewitt, Stephan Glander "Rubber, 5. Solution Rubbers" in Ullmann's Encyclopedia of Industrial Chemistry, 2011, Wiley-VCH, Weinheim. doi:10.1002/14356007.o23_o02
- ↑ Baskara, D.; Muller, A.H.. «Anionic Vinyl Polymerization». A: Controlled and living polymerizations: From mechanisms to applications. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA,, 2010. DOI 10.1002/9783527629091.ch1. ISBN 9783527629091.
- ↑ Bailey, W.F.; etal «Preparation and facile cyclization of 5-alkyn-1-yllithiums». Tetrahedron Lett., 30, 1989, pàg. 3901–3904. DOI: 10.1016/S0040-4039(00)99279-7.
- ↑ 35,0 35,1 35,2 35,3 35,4 35,5 35,6 35,7 Carey, Francis A. «Organometallic compounds of Group I and II metals». A: Advanced Organic Chemistry: Reaction and Synthesis Pt. B. Kindle. Springer, 2007. ISBN 978-0-387-44899-2.
- ↑ Ashby, E.C.; Noding, S.R. «The effects of added salts on the stereoselectivity and rate of organometallic compound addition to ketones». J. Org. Chem., 44, 1979, pàg. 4371–4377. DOI: 10.1021/jo01338a026.
- ↑ Yamataka, Hiroshi. «Addition of organolithium reagents to double bonds». A: PATAI'S Chemistry of Functional Groups.. John Wiley & Sons, Ltd, 2009. DOI 10.1002/9780470682531.pat0310. ISBN 9780470682531.
- ↑ Landa, S.; etal «Über adamantan und dessen derivate IX. In 2-stellung substituierte derivate». Czech. Chem. Commun., 72, 1967, pàg. 570–575. DOI: 10.1135/cccc19670570.
- ↑ Rubottom, G.M.; Kim, C «Preparation of methyl ketones by the sequential treatment of carboxylic acids with methyllithium and chlorotrimethylsilane». J. Org. Chem., 48, 1983, pàg. 1550–1552. DOI: 10.1021/jo00157a038.
- ↑ Zadel, G.; Breitmaier, E. «A One-Pot Synthesis of Ketones and Aldehydes from Carbon Dioxide and Organolithium Compounds». Angew. Chem. Int. Ed., 31, 1992, pàg. 1035–1036. DOI: 10.1002/anie.199210351.
- ↑ Ronald, R.C. «Methoxymethyl ethers. An activating group for rapid and regioselective metalation». Tetrahedron Lett., 16, 1975, pàg. 3973–3974. DOI: 10.1016/S0040-4039(00)91212-7.
- ↑ Hunt, D.A. «Michael addition of organolithium compounds. A Review,». Org. Prep. Proc. Int., 21, 1989, pàg. 705–749. DOI: 10.1080/00304948909356219.
- ↑ Reich, H. J.; Sikorski, W. H. «Regioselectivity of Addition of Organolithium Reagents to Enones: The Role of HMPA». J. Org. Chem., 64, 1999, pàg. 14–15. DOI: 10.1021/jo981765g.
- ↑ Collum, D.B.; etal «NMR Spectroscopic Investigations of Mixed Aggregates Underlying Highly Enantioselective 1,2-Additions of Lithium Cyclopropylacetylide to Quinazolinones». J. Am. Chem. Soc., 123, 2001, pàg. 9135–9143. DOI: 10.1021/ja0105616.
- ↑ Sommmer, L.H.; Korte, W. D. «Stereospecific coupling reactions between organolithium reagents and secondary halides». J. Org. Chem., 35, 1970, pàg. 22–25. DOI: 10.1021/jo00826a006.
- ↑ 46,0 46,1 46,2 46,3 "Organolithium Reagents Reich, H.J. 2002 http://www.chem.wisc.edu/areas/reich/handouts/lireagents/orgli-primer.pdf
- ↑ 47,0 47,1 47,2 47,3 The Preparation of Organolithium Reagents and Intermediates Leroux.F., Schlosser. M., Zohar. E., Marek. I., Wiley, New York. 2004. ISBN 978-0-470-84339-0
- ↑ 48,0 48,1 Hoppe, Dieter; Christoph, Guido. «Asymmetric deprotonation with alkyllithium– (−)-sparteine». A: PATAI'S Chemistry of Functional Groups.. John Wiley & Sons, Ltd, 2009. DOI 10.1002/9780470682531.pat0313. ISBN 9780470682531.
- ↑ Clayden, Jonathan. «Directed metallization of aromatic compounds». A: PATAI'S Chemistry of Functional Groups.. John Wiley & Sons, Ltd, 2009. DOI 10.1002/9780470682531.pat0306. ISBN 9780470682531.
- ↑ Schlosser, M «Superbases for organic synthesis». Pure Appl. Chem., 60, 1988, pàg. 1627–1634. DOI: 10.1351/pac198860111627.
- ↑ Roush, W.R.; etal «Enantioselective synthesis using diisopropyl tartrate modified (E)- and (Z)-crotylboronates: Reactions with achiral aldehydes». Tetrahedron Lett., 29, 1988, pàg. 5579–5582. DOI: 10.1016/S0040-4039(00)80816-3.
- ↑
Park, Y.S.; etal «(−)-Sparteine-Mediated
α -Lithiation of N-Boc-N-(p-methoxyphenyl)benzylamine: Enantioselective Syntheses of (S) and (R) Mono- and Disubstituted N-Boc-benzylamines». J. Am. Chem. Soc., 118, 1996, pàg. 3757–3758. DOI: 10.1021/ja9538804. - ↑ 53,0 53,1 53,2 53,3 Valnot, Jean-Yves; Maddaluno, Jacques. «Aspects of the synthesis, structure and reactivity of lithium enolates». A: PATAI'S Chemistry of Functional Groups.. John Wiley & Sons, Ltd, 2009. DOI 10.1002/9780470682531.pat0345. ISBN 9780470682531.
- ↑ Ireland. R. E.; etal «The ester enolate Claisen rearrangement. Stereochemical control through stereoselective enolate formation». J. Am. Chem. Soc., 98, 1976, pàg. 2868–2877. DOI: 10.1021/ja00426a033.
- ↑ Gilman, Henry.; Langham, Wright.; Jacoby, Arthur L. «Metalation as a Side Reaction in the Preparation of Organolithium Compounds». Journal of the American Chemical Society, 61, 1, 1939, pàg. 106–109. DOI: 10.1021/ja01870a036. ISSN: 0002-7863.
- ↑ Bailey, W. F.; Patricia, J. F. «The mechanism of the lithium - halogen Interchange reaction : a review of the literature». J. Organomet. Chem., 352, 1988, pàg. 1–46. DOI: 10.1016/0022-328X(88)83017-1.
- ↑ 57,0 57,1 Farnham, W. B.; Calabrese, J. C. «Novel hypervalent (10-I-2) iodine structures». J. Am. Chem. Soc, 108, 1986, pàg. 2449–2451. DOI: 10.1021/ja00269a055.
- ↑ Rogers, H. R.; Houk, J. «Preliminary studies of the mechanism of metal-halogen exchange. The kinetics of reaction of n-butyllithium with substituted bromobenzenes in hexane solution». J. Am. Chem. Soc., 104, 1982, pàg. 522–525. DOI: 10.1021/ja00366a024.
- ↑ Fischer, H. «Electron spin resonance of transient alkyl radicals during alkyllithium-alkyl halide reactions». J. Phys. Chem., 73, 1969, pàg. 3834–3838. DOI: 10.1021/j100845a044.
- ↑ Bailey, W.F.; etal «Metal—halogen interchange between t-butyllithium and 1-iodo-5-hexenes provides no evidence for single-electron transfer». Tetrahedron Lett., 27, 1986, pàg. 1861–1864. DOI: 10.1016/s0040-4039(00)84395-6.
- ↑ Seebach, D; Neumann H. «Stereospecific preparation of terminal vinyllithium derivatives by Br/Li-exchange with t-butyllithium». Tetrahedron Lett., 52, 1976, pàg. 4839–4842. DOI: 10.1016/s0040-4039(00)78926-x.
- ↑ Toth, J. E.; Hamann, P.R.; Fuchs, P.L. «Studies culminating in the total synthesis of (dl)-morphine». J. Org. Chem., 53, 1988, pàg. 4694–4708. DOI: 10.1021/jo00255a008.
- ↑ Parham, W.P.; Bradsher, C.K. «Aromatic organolithium reagents bearing electrophilic groups. Preparation by halogen-lithium exchange». Acc. Chem. Res., 15, 1982, pàg. 300–305. DOI: 10.1021/ar00082a001.
- ↑ Sotomayor, N.; Lete, E. «Aryl and Heteroaryllithium Compounds by Metal - Halogen Exchange. Synthesis of Carbocyclic and Heterocyclic Systems». Curr. Org. Chem., 7, 2003, pàg. 275–300. DOI: 10.2174/1385272033372987.
- ↑ Quin, C.; etal «Synthesis of a mitochondria-targeted spin trap using a novel Parham-type cyclization». Tetrahedron, 65, 2009, pàg. 8154–8160. DOI: 10.1016/j.tet.2009.07.081.
- ↑ Corey, E.J.; Wollenberg, R.H. «Useful new organometallic reagents for the synthesis of allylic alcohols by nucleophilic vinylation». J. Org. Chem, 40, 1975, pàg. 2265–2266. DOI: 10.1021/jo00903a037.
- ↑ Reeder, M.R.; etal «An Improved Method for the Palladium Cross-Coupling Reaction of Oxazol-2-ylzinc Derivatives with Aryl Bromides». Org. Process Res. Dev., 7, 2003, pàg. 696–699. DOI: 10.1021/op034059c.
- ↑ Nakamura, E.; etal «Reaction Pathway of the Conjugate Addition of Lithium Organocuprate Clusters to Acrolein». J. Am. Chem. Soc., 119, 1997, pàg. 4900–4910. DOI: 10.1021/ja964209h.
- ↑ "Organometallics in Organic Synthesis", Schlosser, M., Ed, Wiley: New York, 1994. ISBN 0-471-93637-5
- ↑ Si-Fodil, M.; etal «Obtention of 2,2-(diethoxy) vinyl lithium and 2-methyl-4-ethoxy butadienyl lithium by arene-catalysed lithiation of the corresponding chloro derivatives. Synthetic applications». Tetrahedron Lett., 39, 1998, pàg. 8975–8978. DOI: 10.1016/S0040-4039(98)02031-0.
- ↑ Cohen, T; Bhupathy. M «Organoalkali compounds by radical anion induced reductive metalation of phenyl thioethers». Acc. Chem. Res., 22, 1989, pàg. 152–161. DOI: 10.1021/ar00160a006.
- ↑ 72,0 72,1 Snieckus, V «Directed ortho metalation. Tertiary amide and O-carbamate directors in synthetic strategies for polysubstituted aromatics». Chem. Rev., 90, 1990, pàg. 879–933. DOI: 10.1021/cr00104a001.
- ↑ 73,0 73,1 Schwindeman, James A.; Woltermann, Chris J.; Letchford, Robert J. «Safe handling of organolithium compounds in the laboratory». Chemical Health and Safety, 9, 3, 2002, pàg. 6–11. DOI: 10.1016/S1074-9098(02)00295-2. ISSN: 1074-9098.
- ↑ Gellert, H; Ziegler, K. «Organoalkali compounds. XVI. The thermal stability of lithium alkyls.». Liebigs Ann. Chem., 567, 1950, pàg. 179–185. DOI: 10.1002/jlac.19505670110.
- ↑ Kofron, W.G.; Baclawski, L.M. «A convenient method for estimation of alkyllithium concentrations». J. Org. Chem., 41, 1976, pàg. 1879–1880. DOI: 10.1021/jo00872a047.
- ↑ «Directed Ortho-Lithiation of Phenylcarbamic Acid 1,l-Dimethylethyl Ester (N-Boc-aniline). Revision and Improvements». J. Org. Chem., 57, 25, 1992, pàg. 6833–6837. DOI: 10.1021/jo00051a030.
Compostos de carboni amb altres elements de la taula periòdica | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||