
a non-profit association
rDNA ITS based identification of Eukaryotes and their communication via DOIs
Current version: 10.0; Last updated: 2024-04-22 (read more)
Number of ITS sequences (UNITE+INSD): 3 846 536;
Number of UNITE fungal Species Hypotheses with DOIs at 1.5% threshold: 195 696 (more statistics)
UNITE is a database and sequence management environment centered on the eukaryotic nuclear ribosomal ITS region. All eukaryotic ITS sequences from the International Nucleotide Sequence Database Collaboration are clustered to approximately the species level (distance between species in steps of 0.5%), and all such species hypotheses are given a DOI to facilitate unambiguous scientific communication and data assembly. The user can interact with these species hypotheses (e.g., SH1566366.08FU) in various ways, and third-party sequence and metadata annotation are supported.... Registration is needed to access the more powerful features of the accompanying PlutoF workbench. A range of search and query options are available, and precompiled reference datasets for local sequence similarity searches and HTS pipelines are available for download. Owing to the mycological roots of UNITE, two versions of the release files are available: all eukaryotes (including fungi), and fungi only. UNITE is a community effort that relies on the scientific expertise of its users – please consider contributing by, e.g., annotating sequences of your taxa of expertise.
Read more
UNITE Species Hypotheses
UNITE provides unified way how you delimit, identify, communicate and work with DNA based Species Hypotheses (SH). All SHs are connected to the taxon name and classification. Read Kõljalg et al. 2013 paper for the description of the system.
After implementing Species Hypotheses (SH) approach in 2012 UNITE started to assign unique version numbers when new clusters of SHs were computed. Earlier versions are stored in the system and can be revisited for the browsing as well as for the reproducing identifications (but not for the annotating or any other changes). Each SH code includes version number and therefore can be resolved even if the code was published years ago.
×
What is Species Hypothesis?
Species Hypothesis - any species level group of individuals that share a given set of observed characters. Remark: This is a practical definition of the SH which is major building block of the UNITE new system.
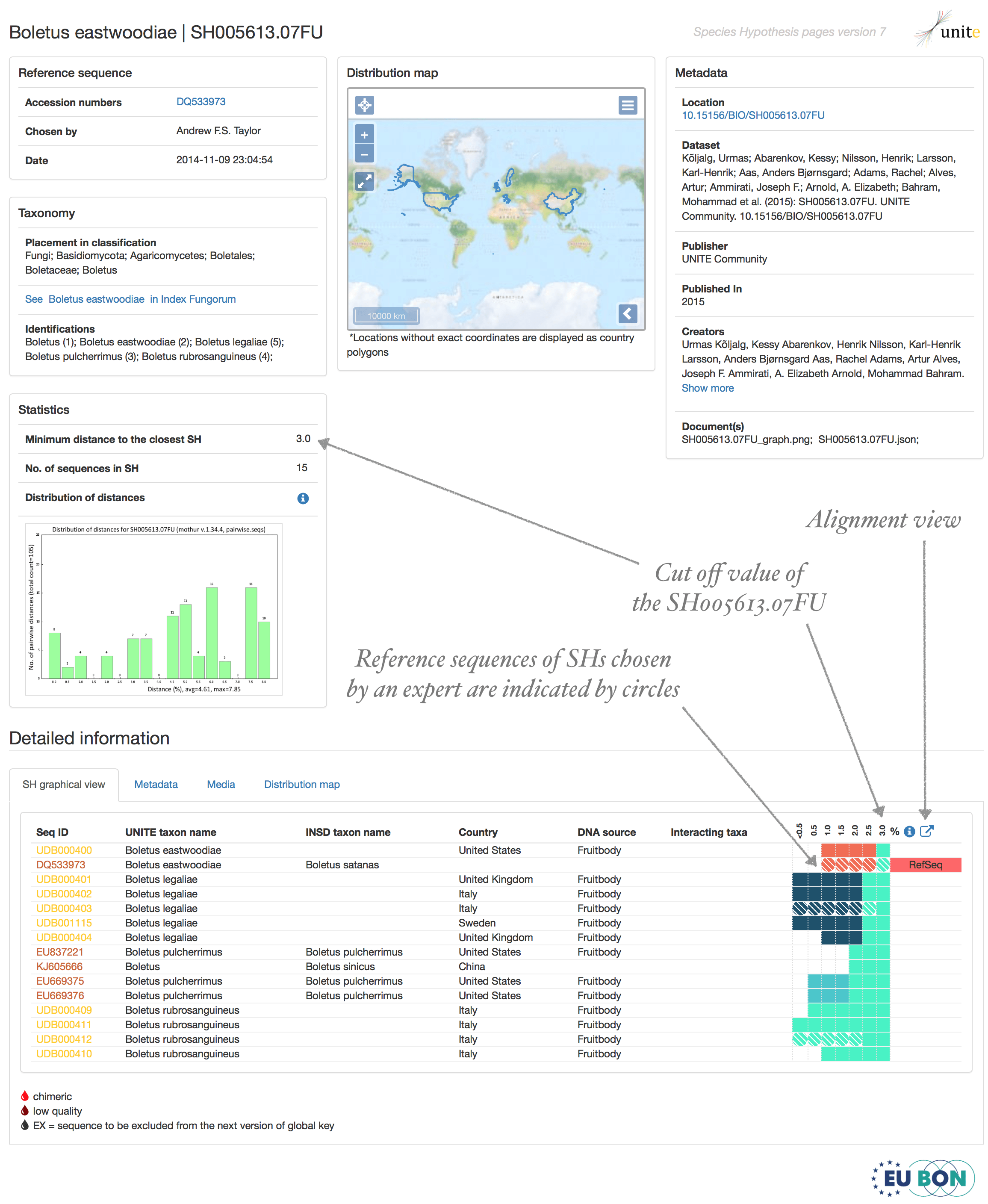
Accession code of the SH - each SH has unique accession code, e.g. SH001616.07FU. Where SH means Species Hypothesis followed by number, full stop, version number and taxon acronym (FU designates for Fungi).
Read how SHs and compound clusters of the closely related SHs are calculated in version 5 here.
 ×
×
What are reference and representative sequences?
Reference sequence (RefS) – serves as a name anchor for the Species Hypothesis and is chosen manually by an expert to define the SH. It may originate from any biological sample, viz. herbarium specimen, living culture, soil, water, air, tissue of other organism, etc. The RefS forms a part of the name of the full name of the SH is thus used for scientific communication.
Accession code of the reference sequence - each SH has a unique accession code - e.g. SH001616.07FU - where SH translates into Species Hypothesis and is followed by a unique number, full stop, version number, and taxon acronym (FU for Fungi).
Representative sequence (RepS) - when RefS are not available (has not been designated manually), a representative sequence (RepS) is chosen automatically from the most common sequence type in the SH. All SHs have a RepS. For SHs for which a RefS has been designated, the RefS takes precedence over the RepS. The RepS are shown in green font in the SH pages (e.g., https://unite.ut.ee/sh/SH001616.07FU).
×
How to submit specimens for sequencing?
Random text here
×
Select distance and dataset to browse, or open taxon name on the tree:
- Threshold - each threshold value shows minimum distance between two sister Species Hypotheses (SH). Threshold value 1.0% means that two sequences belong into different SHs if their ITS2 sequences are more than 1% different. However, they fall into single SH under 1% threshold value if third sequence, which is 0.5% different from both sequences, becomes available. In other words, two sister SHs fall into single SH under specific threshold value if the distance between two most similar sequences in sister SHs are more similar than this specific threshold value requires.
- Include - defines whether to include SH-s based on one sequence only or not. Default value includes both.
- Taxon name - specifying taxon name allows direct entry to the selected taxon node on the tree.
- display this info
- display list of SH-s belonging to selected taxon
- open SH graph in a new window
×
