医薬品 情報
| ナルフラフィン |
|
| ナルフラフィン |
|
| Nalfurafine Hydrochloride | |
| ナルフラフィン |
|
| 1190 | |
| ATCコード | V03AX02 |
| KEGG DRUG |
D06405
ナルフラフィン
|
| JAPIC |
|
添付 文書 情報 2023年 10月 改訂 (第 2版 )

|
2. |
商品 情報
3.組成 ・性状
| YJコード | |||||
|---|---|---|---|---|---|
|
ナルフラフィン |
Nalfurafine Hydrochloride OD Film | ニプロ | 1190015F1054 | 261.4 |
2. 禁忌
2.1 本 剤 の成分 に対 し過敏 症 の既往 歴 のある患者
4. 効能 または効果
○透析 患者
○慢性 肝 疾患 患者
6. 用法 及 び用量
7. 用法 及 び用量 に関連 する注意
<効能 共通 >
<血液 透析 患者 におけるそう痒 症 の改善 の場合 >
7.2 本 剤 の投与 から血液 透析 開始 までは十分 な間隔 をあけること。本 剤 は血液 透析 により除去 されることから、本 剤 服用 から血液 透析 までの時間 が短 い場合 、本 剤 の血 中 濃度 が低下 する可能 性 がある。[16.8.1参照 ]
<腹膜 透析 患者 におけるそう痒 症 の改善 の場合 >
<慢性 肝 疾患 患者 におけるそう痒 症 の改善 の場合 >
7.4 本 剤 の投与 は1日 1回 2.5μ gから開始 し、効果 不十分 な場合 に1日 1回 5μ gへの増量 を検討 すること。
8. 重要 な基本 的 注意
8.1 重度 (Child-Pugh分類 グレードC)の肝 障害 のある患者 に対 する本 剤 の投与 にあたっては、リスク・ベネフィットを勘案 し、投与 中 は患者 の状態 を十分 に観察 すること。[9.3.1、16.1.1参照 ]
8.2 眠気 、めまい等 があらわれることがあるので、本 剤 投与 中 の患者 には自動車 の運転 等 危険 を伴 う機械 の操作 には従事 させないよう注意 すること。
8.3 本 剤 の使用 により効果 が認 められない場合 には、漫然 と長期 にわたり投与 しないように注意 すること。
8.4 本 剤 の投与 により、プロラクチン値 上昇 等 の内分泌 機能 異常 があらわれることがあるので、適宜 検査 を実施 することが望 ましい。
9. 特定 の背景 を有 する患者 に関 する注意
9.2 腎 機能 障害 患者
<慢性 肝 疾患 患者 におけるそう痒 症 の改善 の場合 >
9.3 肝 機能 障害 患者
<効能 共通 >
9.3.1 重度 (Child-Pugh分類 グレードC)の肝 障害 のある患者
<透析 患者 におけるそう痒 症 の改善 の場合 >
9.3.2 中等 度 (Child-Pugh分類 グレードB)の肝 障害 のある患者
9.5 妊 婦
9.6 授乳 婦
9.7 小児 等
9.8 高齢 者
10. 相互 作用
CYP3A4
10.2 併用 注意
| CYP3A4 アゾール [16.7.1、16.7.2 | CYP3A4 | |
| オピオイド |
11. 副作用
11.1 重大 な副作用
11.1.1 肝 機能 障害 、黄疸 (いずれも頻度 不明 )
AST、ALT、Al-P、γ -GTPの著 しい上昇 等 を伴 う肝 機能 障害 、黄疸 があらわれることがある。
11.2 その他 の副作用
| 5% | 1〜5% | 1% | ||
| いらいら | ||||
| そう | ||||
| AST | LDH | |||
| プロラクチン | テストステロン | |||
| その |
13. 過 量 投与
13.1 症状
13.2 処置
14. 適用 上 の注意
14.1 薬剤 交付 時 の注意
14.2 服用 時 の注意
14.2.2 本 剤 は寝 たままの状態 では、水 なしで服用 させないこと。
14.2.3 アルミ包装 で遮光 して品質 を保持 しているため、アルミ包装 開封 後 は速 やかに服用 すること。
15. その他 の注意
15.2 非 臨床 試験 に基 づく情報
15.2.1 動物 実験 (イヌ静脈 内 投与 、0.1μ g/kg以上 )において全身 麻酔 下 での血圧 低下 が報告 されている。
15.2.2 動物 実験 (ラット筋肉 内 投与 、40μ g/kg/day以上 )において受胎 率 の低下 が報告 されている。
16. 薬物 動態
16.1 血 中 濃度
16.1.1 単 回 投与
(1)血液 透析 患者 (16例 )にナルフラフィン塩酸 塩 (カプセル)2.5又 は5μ gを経口 単 回 投与 した時 、未 変化 体 の薬物 動態 パラメータは以下 の通 りであった1)。
| Cmax(pg/mL) | Tmax(hr) | AUC0-∞(pg・hr/mL) | t1/2(hr) | ||
| 2.5 | 8 | 3.15±0.82 | 4.25±1.58 | 66.26±15.54※ | 14.21±4.93※ |
| 5 | 8 | 6.51±2.76 | 3.00±0.93 | 120.59±71.90 | 14.03±7.44 |
(2)腹膜 透析 患者 (16例 )にナルフラフィン塩酸 塩 (カプセル)2.5又 は5μ gを経口 単 回 投与 した時 、未 変化 体 の薬物 動態 パラメータは以下 の通 りであった。腹膜 透析 の方法 (連続 携行 式 腹膜 透析 (CAPD)、持続 的 周期 的 腹膜 透析 (CCPD))、自動 腹膜 潅流装置 (APD)の有無 及 び透析 液 の種類 により、未 変化 体 の薬物 動態 パラメータに明 らかな差異 は認 められなかった。なお、ナルフラフィン塩酸 塩 (カプセル)5μ g投与 群 において、ナルフラフィン塩酸 塩 (カプセル)投与 から初回 の透析 液 交換 までの時間 が3時 間 と規定 された5例 のうち1例 で、未 変化 体 のCmax及 びAUC0-∞がそれぞれ5.37pg/mL及 び156.54pg・h/mLと低下 する傾向 が認 められた2)。[7.3参照 ]
| Cmax(pg/mL) | Tmax(hr)※ | AUC0-∞(pg・hr/mL) | t1/2(hr) | ||
| 2.5 | 5 | 3.81±0.88 | 1.00 | 92.67±23.47 | 20.99±4.22 |
| 5 | 11 | 8.28±3.00 | 2.00 | 193.74±57.52 | 24.77±3.23 |
(3)軽度 (Child-Pugh分類 グレードA)の代償 性 肝硬変 患者 (12例 )にナルフラフィン塩酸 塩 (カプセル)2.5又 は5μ gを経口 単 回 投与 した時 、未 変化 体 の薬物 動態 パラメータは以下 の通 りであった。健康 成人 男子 と比較 してCmaxやAUCが上昇 する傾向 は認 められなかった3)。
| Cmax(pg/mL) | Tmax(hr) | AUC0-∞(pg・hr/mL) | t1/2(hr) | ||
| 2.5 | 6 | 3.63±1.26 | 2.33±1.03 | 34.58±13.55※ | 5.37±2.11※ |
| 5 | 6 | 6.76±2.03 | 1.50±0.55 | 58.06±26.28 | 6.61±2.46 |
(4)中等 度 (Child-Pugh分類 グレードB)の慢性 肝 疾患 患者 (延 べ30例 )にナルフラフィン塩酸 塩 (カプセル)2.5又 は5μ gを経口 単 回 投与 した時 、未 変化 体 の薬物 動態 パラメータは以下 の通 りであった。軽度 (Child-Pugh分類 グレードA)の肝 障害 患者 と比較 してCmaxとAUCは上昇 する傾向 が認 められた4)。[9.3.2参照 ]
| Cmax(pg/mL) | Tmax(hr) | AUC0-∞(pg・hr/mL) | t1/2(hr) | ||
| 2.5 | 16 | 6.36±2.62 | 1.81±1.52 | 117.4±51.4 | 17.52±10.69 |
| 5 | 14 | 11.71±4.45 | 1.50±1.02 | 197.7±97.0 | 14.59±5.27 |
16.1.2 反復 投与
| Cmax(pg/mL) | Tmax(hr) | AUC0-∞(pg・hr/mL) | t1/2(hr) | ||
| 2.5 | 7 | 5.70±3.85 | 4.14±1.35 | 210.25±144.28※ | 25.33±10.52※ |
| 5 | 7 | 10.25±1.74 | 3.86±1.21 | 358.86±179.24 | 28.34±8.55 |
また、透析 時 では非 透析 時 と比較 しt1/2が短縮 しており、透析 時 及 び非 透析 時 のt1/2はそれぞれ、5.11〜11.17(hr)、13.55〜64.37(hr)であった。
16.1.3 生物 学 的 同等 性 試験
ナルフラフィン塩酸 塩 ODフィルム2.5μ g「ニプロ」とレミッチカプセル2.5μ gのそれぞれ1枚 又 は1カプセル(ナルフラフィン塩酸 塩 として2.5μ g)を、クロスオーバー法 により健康 成人 男子 に絶食 単 回 経口 投与 して血漿 中 ナルフラフィン濃度 を測定 した。得 られた薬物 動態 パラメータ(AU C0→48hr、Cmax)について90%信頼 区間 法 にて統計 解析 を行 った結果 、log(0.80)〜log(1.25)の範囲 内 であり、両剤 の生物 学 的 同等 性 が確認 された6)7)。
血漿 中 ナルフラフィン濃度 推移 (水 あり投与 )
血漿 中 ナルフラフィン濃度 推移 (水 なし投与 )
| AUC0→48hr(pg・hr/mL) | Cmax(pg/mL) | Tmax(hr) | t1/2(hr) | |
| ナルフラフィン | 28.904±5.634 | 2.2638±0.4896 | 2.68±0.75 | 9.35±1.41 |
| レミッチカプセル2.5 | 28.060±5.070 | 2.3160±0.4942 | 2.53±0.70 | 8.95±1.73 |
| AUC0→48hr(pg・hr/mL) | Cmax(pg/mL) | Tmax(hr) | t1/2(hr) | |
| ナルフラフィン | 28.520±5.315 | 2.1440±0.4159 | 2.83±0.86 | 9.35±1.98 |
| レミッチカプセル2.5 | 29.475±6.168 | 2.4655±0.5025 | 2.28±1.02 | 9.38±1.85 |
16.2 吸収
16.2.1 食事 の影響
(注 1)通常 1回 投与 量 は2.5μ gである。
(注 2)開発 段階 の製剤 での試験 成績 であるが、当該 製剤 はレミッチカプセルと溶出 挙動 の類似 性 から同等 であると考 えられている9)。
| Cmax(pg/mL) | Tmax(hr) | AUC0-48hr(pg・hr/mL) | t1/2(hr) | |
| 12.67±3.95 | 3.1±1.1 | 114.46±34.26 | 5.99±1.35 | |
| 13.68±3.65 | 3.2±1.3 | 126.03±38.10 | 5.90±1.10 |
16.3 分布
16.3.2 ラットに経口 単 回 投与 した後 の全身 オートラジオグラム及 び組織 中 放射能 濃度 測定 結果 から、投与 後 15分 に食道 、肝臓 、消化 管 及 びその内容 物 に高 い放射能 の分布 が認 められた。また、投与 後 168時 間 では肝臓 、腎臓 、甲状腺 及 び腸 内容 物 に放射能 が認 められた10)。
16.4 代謝
16.5 排泄
16.5.1 健康 成人 男子 (6例 )を対象 に、トリチウムで標識 したナルフラフィン塩酸 塩 を静脈 内 単 回 投与 した時 の薬物 動態 を検討 したところ、投与 後 14日間 での糞 中 排泄 率 は56.0%、尿 中 の排泄 率 は36.2%で、累積 排泄 率 は92.2%となった。尿 中 では主 に未 変化 体 として、糞 中 では主 に脱 シクロプロピルメチル体 として排泄 された12)。主 代謝 物 は脱 シクロプロピルメチル体 であり、その他 にグルクロン酸 抱合 体 が認 められた(外国 人 データ)。
16.5.2 4種 の透析 膜 を用 いて透析 による除去 について検討 したところ、未 変化 体 の透析 膜 面積 1.5m2換算 クリアランスは44.6〜61.8mL/minと算出 され、健康 成人 男子 における未 変化 体 の腎 クリアランス170〜210mL/minと比較 すると小 さいものの、未 変化 体 は膜 種 に関係 なく透析 により除去 されるものと考 えられた。また、代謝 物 (脱 シクロプロピルメチル体 及 びグルクロン酸 抱合 体 )についても膜 種 に関係 なく除去 されるものと考 えられた10)。
16.7 薬物 相互 作用
16.7.1 ケトコナゾール(経口 剤 :国内 未 発売 )との併用
(注 )通常 1回 投与 量 は2.5μ gである。
16.7.2 in vitro試験 、代謝
16.7.3 ヒトP糖 タンパク(MDR1)発現 LLC-PK1細胞 を用 いたin vitro試験
16.7.4 非 吸収 性 薬剤 とのin vitro吸着 試験
16.8 その他
17. 臨床 成績
17.1 有効 性 及 び安全 性 に関 する試験
ナルフラフィン塩酸 塩 (カプセル)の成績 を以下 に示 す。既存 治療 抵抗 性 のそう痒 症 を有 する血液 透析 患者 337例 を対象 に、1日 1回 、14日間 経口 反復 投与 した際 の有効 性 を、かゆみの指標 であるVAS(Visual Analogue Scale)を用 い、多 施設 二 重 盲 検 比較 試験 により検討 した。その結果 、投与 前後 でのVAS変化 量 において、2.5μ g及 び5μ g投与 群 で有効 性 が確認 された19)20)。
副作用 発現 率 は、2.5μ g群 で25.0%(28/112例 )、5μ g群 で35.1%(40/114例 )であった。主 な副作用 は、2.5μ g群 で不眠 7.1%(8/112例 )、眠気 4.5%(5/112例 )、便秘 及 びプロラクチン上昇 2.7%(3/112例 )、5μ g群 で不眠 14.0%(16/114例 )、便秘 7.0%(8/114例 )、眠気 3.5%(4/114例 )、そう痒 の悪化 、プロラクチン上昇 及 び甲状腺 刺激 ホルモン上昇 2.6%(3/114例 )であった。
既存 治療 抵抗 性 のそう痒 症 を有 する血液 透析 患者 211例 を対象 に、1日 1回 、ナルフラフィン塩酸 塩 5μ gを52週間 経口 反復 投与 した際 の有効 性 を、VASを用 い、オープン試験 により検討 した。その結果 、投与 前後 でのVAS変化 量 において、有効 性 が確認 された21)。
既存 治療 抵抗 性 のそう痒 症 を有 する腹膜 透析 患者 37例 を対象 に、ナルフラフィン塩酸 塩 2.5μ gを2週間 、続 いて5μ gを2週間 経口 反復 投与 した際 の有効 性 を、かゆみの指標 であるVASを用 い、非 盲 検 非 対照 試験 により検討 した。その結果 、2.5μ g投与 期間 2週 目 (LOCF※)における投与 前後 でのVAS変化 量 の平均 値 は24.93mm(90%信頼 区間 :18.67,31.19mm)であり、90%信頼 区間 の下限 値 は、事前 に設定 されたVAS変化 量 の閾値(15.24mm)を上回 った22)。副作用 発現 率 は、45.9%(17/37例 )であった。主 な副作用 は、不眠 及 びプロラクチン上昇 13.5%(5/37例 )、眠気 及 びテストステロン低下 8.1%(3/37例 )、嘔吐 5.4%(2/37例 )であった。
抗 ヒスタミン薬 又 は抗 アレルギー薬 による治療 が奏効 しない難治 性 のそう痒 症 を有 する慢性 肝 疾患 患者 ※317例 を対象 に、1日 1回 、12週間 経口 反復 投与 した際 の有効 性 を、かゆみの指標 であるVASを用 い、多 施設 二 重 盲 検 比較 試験 により検討 した。主要 評価 項目 は、投与 期間 4週 目 (LOCF)のVAS変化 量 とした。その結果 、投与 前後 でのVAS変化 量 において、2.5μ g及 び5μ g投与 群 で有効 性 が確認 された23)24)。
副作用 発現 率 は、2.5μ g群 で60.0%(63/105例 )、5μ g群 で54.1%(59/109例 )であった。主 な副作用 は、2.5μ g群 でプロラクチン上昇 13.3%(14/105例 )、抗 利尿 ホルモン上昇 及 び総 胆汁 酸 上昇 7.6%(8/105例 )、甲状腺 刺激 ホルモン上昇 6.7%(7/105例 )、不眠症 、頻 尿 ・夜間 頻 尿 及 び眠気 5.7%(6/105例 )、5μ g群 で、頻 尿 ・夜間 頻 尿 、便秘 、眠気 、プロラクチン上昇 及 び抗 利尿 ホルモン上昇 7.3%(8/109例 )、浮動 性 めまい5.5%(6/109例 )であった。
抗 ヒスタミン薬 又 は抗 アレルギー薬 による治療 が奏効 しない難治 性 のそう痒 症 を有 する慢性 肝 疾患 患者 ※122例 を対象 に、1日 1回 、ナルフラフィン塩酸 塩 5μ gを52週間 経口 反復 投与 した際 の有効 性 を、VASを用 い、オープン試験 により検討 した。その結果 、投与 前後 でのVAS変化 量 において有効 性 が確認 された25)。
<血液 透析 患者 におけるそう痒 症 の改善 の場合 >
17.1.1 国内 第 III相 試験
| プラセボ [95% | p | ||||
| プラセボ | 111 | 73.78±11.47 | 58.55±22.06 | 9.13[3.78,14.49] | p=0.0005 |
| 2.5 | 112 | 76.71±11.79 | 52.19±23.71 | ||
| プラセボ [95% | p | ||||
| プラセボ | 111 | 73.78±11.47 | 58.55±22.06 | 8.26[3.05,13.47] | p=0.0010 |
| 5 | 114 | 73.03±11.54 | 49.63±22.30 | ||
※投与 前 の平均 VAS値 を共 変量 とした共 分散 分析 により調整 済 みの点 推定 値
17.1.2 国内 第 III相 試験 (長期 投与 試験 )
| 2 | 4 | 12 | 24 | 36 | 52 | ||
| 211 | 208 | 198 | 184 | 163 | 155 | 145 | |
| 75.22±12.41 | 50.95±24.38 | 47.17±25.32 | 39.39±25.83 | 33.60±27.73 | 31.85±24.91 | 30.87±25.92 |
ナルフラフィン塩酸 塩 の依存 性 について、精神 依存 及 び身体 依存 を示 す症例 は認 められなかった。また耐 性 が211例 中 5例 に認 められている21)。
副作用 発現 率 は、48.8%(103/211例 )であった。主 な副作用 は、不眠症 19.4%(41/211例 )、便秘 7.1%(15/211例 )、プロラクチン上昇 3.3%(7/211例 )、眠気 2.4%(5/211例 )であった。
<腹膜 透析 患者 におけるそう痒 症 の改善 の場合 >
17.1.3 国内 第 III相 試験
※LOCF:Last Observation Carried Forward
<慢性 肝 疾患 患者 におけるそう痒 症 の改善 の場合 >
17.1.4 国内 第 III相 試験
※原 疾患 が確定 しており、肝臓 の炎症 が6ヵ月 以上 持続 している又 は画像 診断 により肝炎 からさらに病態 が進展 した状態 にあると判断 された肝 疾患 患者
| プラセボ [95% | p | ||||
| プラセボ | 103 | 77.26±10.50 | 58.02±24.11 | 9.31[2.94,15.69] | p=0.0022 |
| 2.5 | 105 | 77.30±11.04 | 48.74±25.27 | ||
| プラセボ [95% | p | ||||
| プラセボ | 103 | 77.26±10.50 | 58.02±24.11 | 8.22※※※[1.88,14.55] | p=0.0056 |
| 5 | 109 | 77.29±11.07 | 49.79±25.50※※※ | ||
※※投与 前 の平均 VAS値 を共 変量 とした共 分散 分析 により調整 済 みの点 推定 値
※※※108例
17.1.5 国内 第 III相 試験 (長期 投与 試験 )
※原 疾患 が確定 しており、肝臓 の炎症 が6ヵ月 以上 持続 している又 は画像 診断 により肝炎 からさらに病態 が進展 した状態 にあると判断 された肝 疾患 患者
| 2 | 4 | 12 | 24 | 36 | 52 | ||
| 122 | 122 | 121 | 116 | 110 | 103 | 99 | |
| 78.05±11.73 | 56.70±24.57 | 50.09±26.94 | 42.88±28.61 | 37.67±27.23 | 31.31±25.43 | 27.77±24.73 |
ナルフラフィン塩酸 塩 の依存 性 について、精神 依存 を示 す症例 は認 められなかった。また、122例 中 、身体 依存 が1例 、耐 性 が4例 に認 められている25)。
副作用 発現 率 は、75.4%(92/122例 )であった。主 な副作用 は、頻 尿 ・夜間 頻 尿 13.1%(16/122例 )、プロラクチン上昇 11.5%(14/122例 )、便秘 10.7%(13/122例 )、浮動 性 めまい7.4%(9/122例 )、抗 利尿 ホルモン上昇 6.6%(8/122例 )、総 胆汁 酸 上昇 5.7%(7/122例 )であった。
18. 薬効 薬理
18.1 作用 機 序
ヒトオピオイド受容 体 発現 細胞 を用 いたin vitroの受容 体 結合 試験 及 び受容 体 作動 性 試験 の結果 から、選択 的 なオピオイドκ 受容 体 作動 薬 であることが示 されている26)。
Ki | 0.244±0.0256 | 2.21±0.214 | 484±59.6 | 1:9:1984 |
EC50(nmol/L) | 0.00816±0.00138 | 1.66±0.09 | 21.3±1.0 | 1:203:2610 |
18.2 そう痒 に対 する作用
18.3 依存 性
19. 有効 成分 に関 する理化学 的 知見
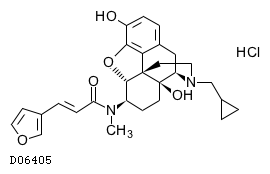
19.1. ナルフラフィン塩酸 塩
| ナルフラフィン |
|
| Nalfurafine Hydrochloride | |
| (2E)-N-[(5R,6R)-17-(Cyclopropylmethyl)-4,5-epoxy-3,14-dihydroxymorphinan-6-yl]-3-(furan-3-yl)-N-methylprop-2-enamide monohydrochloride | |
| C28H32N2O5・HCl | |
| 513.03 | |
| KEGG DRUG |
|
22. 包装
14枚 、140枚
23. 主要 文献
-
血液 透析 患者 における薬物 動態 の検討 (レミッチカプセル:2009年 1月 21日 承認 、CTD2.7.6) -
腹膜 透析 患者 における薬物 動態 の検討 (レミッチカプセル・OD錠 、ノピコールカプセル:2017年 9月 22日 承認 、審査 報告 書 ) -
代償 性 肝硬変 患者 における薬物 動態 の検討 (ノピコールカプセル:2014年 12月26日 承認 、CTD2.7.6.7) -
Child-Pugh
分類 グレードBの肝硬変 患者 における薬物 動態 の検討 (ノピコールカプセル:2014年 12月26日 承認 、CTD2.7.6.8) -
中等 度 以上 の肝 機能 障害 患者 に対 する本 剤 の投与 について(ノピコールカプセル:2014年 12月26日 承認 、審査 報告 書 ) -
社内 資料 :生物 学 的 同等 性 試験 (水 あり) -
社内 資料 :生物 学 的 同等 性 試験 (水 なし) -
健康 成人 における食事 の影響 の検討 (レミッチカプセル:2009年 1月 21日 承認 、CTD2.7.6) -
健康 成人 における食事 の影響 の検討 (レミッチカプセル:2009年 1月 21日 承認 、審査 報告 書 ) -
中尾 薫 他 ,日本 薬理 学 雑誌 , 135 (5), 205-214, (2010) »PubMed - Ando,A.et al., Biopharm.Drug Dispos., 33 (5), 257-264, (2012) »PubMed
-
健康 成人 における吸収 、代謝 、排泄 の検討 (レミッチカプセル:2009年 1月 21日 承認 、CTD2.7.6) -
健康 成人 における薬物 相互 作用 の検討 (レミッチカプセル:2009年 1月 21日 承認 、CTD2.7.6), (L20200002) -
薬物 相互 作用 の検討 (レミッチカプセル:2009年 1月 21日 承認 、CTD2.6.4.7), (L20201666) -
ヒトP
糖 タンパク(MDR1)発現 LLC-PK1細胞 を用 いたin vitro試験 (ノピコールカプセル:2014年 12月26日 承認 、CTD2.6.4.7), (L20201667) -
非 吸収 性 薬剤 (吸着 剤 )との薬物 相互 作用 の検討 (ノピコールカプセル:2014年 12月26日 承認 、CTD2.6.4.7) -
非 吸収 性 薬剤 (吸着 剤 )との薬物 相互 作用 の検討 (2)(レミッチカプセル・OD錠 、ノピコールカプセル:2017年 9月 22日 承認 、審査 報告 書 ) -
血液 透析 の影響 (レミッチカプセル:2009年 1月 21日 承認 、審査 報告 書 ) -
血液 透析 患者 におけるそう痒 症 に対 する効果 の検討 (検証 的 試験 )(レミッチカプセル:2009年 1月 21日 承認 、CTD2.7.6) -
血液 透析 患者 におけるそう痒 症 に対 する効果 の検討 (検証 的 試験 )(レミッチカプセル:2009年 1月 21日 承認 、審査 報告 書 ) -
血液 透析 患者 におけるそう痒 症 に対 する効果 の検討 (長期 投与 試験 )(レミッチカプセル:2009年 1月 21日 承認 、CTD2.7.6) -
腹膜 透析 患者 におけるそう痒 症 に対 する効果 の検討 (一般 臨床 試験 )(レミッチカプセル・OD錠 、ノピコールカプセル:2017年 9月 22日 承認 、審査 報告 書 ) -
慢性 肝 疾患 患者 におけるそう痒 症 に対 する効果 の検討 (検証 的 試験 )(ノピコールカプセル:2014年 12月26日 承認 、CTD2.7.6.13) -
慢性 肝 疾患 患者 におけるそう痒 症 に対 する効果 の検討 (検証 的 試験 )(ノピコールカプセル:2014年 12月26日 承認 、審査 報告 書 ) -
慢性 肝 疾患 患者 におけるそう痒 症 に対 する効果 の検討 (長期 投与 試験 )(ノピコールカプセル:2014年 12月26日 承認 、CTD2.7.6.14) -
中尾 薫 他 ,日本 神経 精神 薬理 学 雑誌 , 28 (2), 75-83, (2008) »PubMed -
各種 受容 体 、トランスポーターおよびイオンチャンネルに対 する結合 試験 (ノピコールカプセル:2014年 12月26日 承認 、CTD2.6.2.2) - Umeuchi,H.et al., Eur.J.Pharmacol., 477 (1), 29-35, (2003) »PubMed
- Togashi,Y.et al., Eur.J.Pharmacol., 435 (2-3), 259-264, (2002) »PubMed
24. 文献 請求 先 及 び問 い合 わせ先
ニプロ株式会社
医薬品 情報 室
〒566-8510
大阪 府 摂津 市 千里丘 新町 3番 26号
FAX:050-3535-8939
ニプロ株式会社
医薬品 情報 室
〒566-8510
大阪 府 摂津 市 千里丘 新町 3番 26号
FAX:050-3535-8939
26. 製造 販売 業者 等
26.1 製造 販売元
ニプロ株式会社